
6LU7
In silico evaluation of the compounds of the ayurvedic drug, AYUSH-64, for the action against the SARS-CoV-2 main protease 2

6LU7
Structure-based virtual screening, in silico docking, ADME properties prediction and molecular dynamics studies for the identification of potential inhibitors against SARS-CoV-2 Mpro 3

1PYX
Revisiting the Proposition of Binding Pockets and Bioactive Poses for GSK-3β Allosteric Modulators Addressed to Neurodegenerative Diseases 1
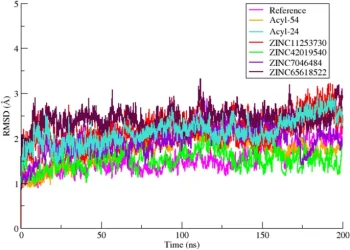
4AF3
Exploration of the structural requirements of Aurora Kinase B inhibitors by a combined QSAR, modelling and molecular simulation approach 1

6lu7
An in-silico evaluation of COVID-19 main protease with clinically approved drugs 1

2AZ5
Structural insight into TNF-α inhibitors through combining pharmacophore-based virtual screening and molecular dynamic simulation 1

4PMI
Probing the mechanism of peptide binding to REV response element RNA of HIV-1; MD simulations and free energy calculations 1

1M48
Molecular dynamics simulation of interleukin-2 and its complex and determination of the binding free energy 1

PDB ID – not available
Theoretical investigation of selective ligand binding mode of galanin receptors

PDB ID – not available
Theoretical investigation of selective ligand binding mode of galanin receptors 2